Active Softwares
Pan1c
![]() Description: Pan1c is a Snakemake workflow that simplifies the creation of pangenomes by focusing on the comparison of similar chromosomes, using either PGGB or Minigraph-Cactus. A pangenome graph is first generated individually for each chromosome, and then these chromosome-scale pangenomes are concatenated into a final graph. This approach reduces construction time through parallelization and facilitates interpretation as well as downstream analyses. Throughout the process, various graphs and statistics are produced to help users assess the quality of the pangenome graph and interpret the results. All these visualizations and statistics are accessible via a web page generated by Pan1c-view.
Description: Pan1c is a Snakemake workflow that simplifies the creation of pangenomes by focusing on the comparison of similar chromosomes, using either PGGB or Minigraph-Cactus. A pangenome graph is first generated individually for each chromosome, and then these chromosome-scale pangenomes are concatenated into a final graph. This approach reduces construction time through parallelization and facilitates interpretation as well as downstream analyses. Throughout the process, various graphs and statistics are produced to help users assess the quality of the pangenome graph and interpret the results. All these visualizations and statistics are accessible via a web page generated by Pan1c-view.
Availability: Pan1c is available on INRAE Forge.
snoBoard
![]() Description: The snoBoard database compiles ribosomal RNA (rRNA) modifications, box C/D and box H/ACA snoRNA guides sequences for a number of species including Saccharomyces cerevisiae (Sc), Human (Hs), the plant Arabidopsis thaliana (At) and tomato Solanum lycopersicum (Sl). Mapped modifications are 2’-O methylation, pseudouridylation and other known modifications when available. The user interface allows the access to rRNA targets, mapped modifications and snoRNA guides under different views. Modifications can be accessed through a table (one line per modification), or mapped on their target RNA sequences and secondary structures. A comparative view allows to access to mapped sites conserved between a selected subset of species. Using the comparative view, it is possible to interrogate and visualize the conservation of target nucleotides sequences and modified sites. Information on modifications targets and snoRNA guides can be accessed by organism, snoRNA guide family and modification type.
Description: The snoBoard database compiles ribosomal RNA (rRNA) modifications, box C/D and box H/ACA snoRNA guides sequences for a number of species including Saccharomyces cerevisiae (Sc), Human (Hs), the plant Arabidopsis thaliana (At) and tomato Solanum lycopersicum (Sl). Mapped modifications are 2’-O methylation, pseudouridylation and other known modifications when available. The user interface allows the access to rRNA targets, mapped modifications and snoRNA guides under different views. Modifications can be accessed through a table (one line per modification), or mapped on their target RNA sequences and secondary structures. A comparative view allows to access to mapped sites conserved between a selected subset of species. Using the comparative view, it is possible to interrogate and visualize the conservation of target nucleotides sequences and modified sites. Information on modifications targets and snoRNA guides can be accessed by organism, snoRNA guide family and modification type.
Availability: Snoboard can be accessed online at https://snoboard.org. The source code is available on INRAE Forge.
ASTERICS
![]() Description: ASTERICS is A Tool for the ExploRation and Integration of omiCS data. It is a web application that aims at making complex exploratory and integration analysis workflows easily available to biologists. Edition, Exploration and Integration menus organize the interface to perform data edition, missing value imputation, normalization, data exploration with interactive plots, numerical summaries, PCA, tests, clustering, self-organizing maps, and data integration with various methods. This project has been funded by Région Occitanie (Grant number 20008788).
Description: ASTERICS is A Tool for the ExploRation and Integration of omiCS data. It is a web application that aims at making complex exploratory and integration analysis workflows easily available to biologists. Edition, Exploration and Integration menus organize the interface to perform data edition, missing value imputation, normalization, data exploration with interactive plots, numerical summaries, PCA, tests, clustering, self-organizing maps, and data integration with various methods. This project has been funded by Région Occitanie (Grant number 20008788).
Availability: ASTERICS can be accessed online at this link and its user documentation (including two case studies) can be accessed here. The source code and its complete installation instruction (using docker images) is available on INRAE Forge.
MetagWGS
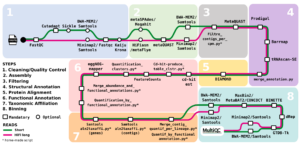 Description: metagWGS is a workflow dedicated to the analysis of metagenomic data. It allows assembly, taxonomic annotation, and functional annotation of predicted genes. Since release 2.3, binning step with the possibility of cross-alignment is included. It has been developed in collaboration with several CATI BIOS4biol agents. Funded by Antiselfish Project (Labex Ecofect), ExpoMicoPig project (France Futur elevage) and SeqOccIn project (CPER – Occitanie Toulouse / FEDER), ATB_Biofilm funded by PNREST Anses, France genomique (ANR-10-INBS-09-08) and Resalab Ouest.
Description: metagWGS is a workflow dedicated to the analysis of metagenomic data. It allows assembly, taxonomic annotation, and functional annotation of predicted genes. Since release 2.3, binning step with the possibility of cross-alignment is included. It has been developed in collaboration with several CATI BIOS4biol agents. Funded by Antiselfish Project (Labex Ecofect), ExpoMicoPig project (France Futur elevage) and SeqOccIn project (CPER – Occitanie Toulouse / FEDER), ATB_Biofilm funded by PNREST Anses, France genomique (ANR-10-INBS-09-08) and Resalab Ouest.
Availability: The source code and its complete documentation is available on INRAE Forge
D-GENIES : Dot plot large GENomes in an interactive, efficient and simple way
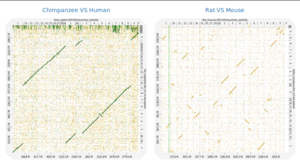 Description: Dot plots are widely used to quickly compare sequence sets. They provide a synthetic similarity overview, highlighting repetitions, breaks and inversions. Different tools have been developed to easily generated genomic alignment dot plots, but they are often limited in the input sequence size. D-GENIES is a standalone and WEB application performing large genome alignments using minimap2 software package and generating interactive dot plots. It enables users to sort query sequences along the reference, zoom in the plot and download several image, alignment or sequence files. D-GENIES is an easy to install open source software package (GPL) developed in Python and JavaScript. The source code is available at https://github.com/genotoul-bioinfo/dgenie and it can be tested at http://dgenies.toulouse.inrae.fr/.
Description: Dot plots are widely used to quickly compare sequence sets. They provide a synthetic similarity overview, highlighting repetitions, breaks and inversions. Different tools have been developed to easily generated genomic alignment dot plots, but they are often limited in the input sequence size. D-GENIES is a standalone and WEB application performing large genome alignments using minimap2 software package and generating interactive dot plots. It enables users to sort query sequences along the reference, zoom in the plot and download several image, alignment or sequence files. D-GENIES is an easy to install open source software package (GPL) developed in Python and JavaScript. The source code is available at https://github.com/genotoul-bioinfo/dgenie and it can be tested at http://dgenies.toulouse.inrae.fr/.
Maintained only Softwares
mixKernel
 Description: Kernel-based methods are powerful methods for integrating heterogeneous types of data. mixKernel provides methods to combine multiple kernels for unsupervised exploratory analysis or to select features in a kernel for unsupervised analysis or kernel-output prediction. For the multiple kernel problem, different solutions have been implemented to obtain a consensus kernel or a kernel that best preserves the original topology of the data. mixKernel also integrates kernel PCA to visualize similarities between samples in a nonlinear space and from the multiple source point of view. Functions to assess and display important variables using permutations are also provided in the package.
Description: Kernel-based methods are powerful methods for integrating heterogeneous types of data. mixKernel provides methods to combine multiple kernels for unsupervised exploratory analysis or to select features in a kernel for unsupervised analysis or kernel-output prediction. For the multiple kernel problem, different solutions have been implemented to obtain a consensus kernel or a kernel that best preserves the original topology of the data. mixKernel also integrates kernel PCA to visualize similarities between samples in a nonlinear space and from the multiple source point of view. Functions to assess and display important variables using permutations are also provided in the package.
Availability: mixKernel is available on CRAN and information are provided on its website. It is fully compatible with the mixOmics package and a tutorial describing the approach can be found on mixOmics web site.
Publications:
- Mariette, J. and Villa-Vialaneix, N. Unsupervised multiple kernel learning for heterogeneous data integration. Bioinformatics. 2017. btx682. [link]
- Brouard, C., Mariette, J., Flamary, R., & Vialaneix, N. Feature selection for kernel methods in systems biology. NAR Genomics and Bioinformatics. 2022. lqac014. [link]
Archived Softwares
sRNAbrowse
 Description: In sRNAseq data sets, only a very small fraction of reads can be assigned to a known functionnal family resulting in a lot of sRNAseq data orphan of functional annotation. The bias introduced by errors, the editing of some sequences but also the lack of similarities in existing ncRNA databases make challenging their structural and functional annotation. sRNAseq data analysis tools such as miRDeep, miRanalyzer and others focus on microRNAs annotation and prediction, neglecting other types of RNAs. Recently, web tools such as DARIO, Ncpro enlarged functional annotation. sRNAbrowse is a software under development which aims at profiling, annotating and exploring as many sRNAseq data as possible, considering different ncRNA families and differential expression in multiple conditions and/or tissues.
Description: In sRNAseq data sets, only a very small fraction of reads can be assigned to a known functionnal family resulting in a lot of sRNAseq data orphan of functional annotation. The bias introduced by errors, the editing of some sequences but also the lack of similarities in existing ncRNA databases make challenging their structural and functional annotation. sRNAseq data analysis tools such as miRDeep, miRanalyzer and others focus on microRNAs annotation and prediction, neglecting other types of RNAs. Recently, web tools such as DARIO, Ncpro enlarged functional annotation. sRNAbrowse is a software under development which aims at profiling, annotating and exploring as many sRNAseq data as possible, considering different ncRNA families and differential expression in multiple conditions and/or tissues.
Availability: sRNAbrowse is freely available on INRAE Forge (workflow srnaseq).
Published instance: Juanchich et al, 2016
jflow
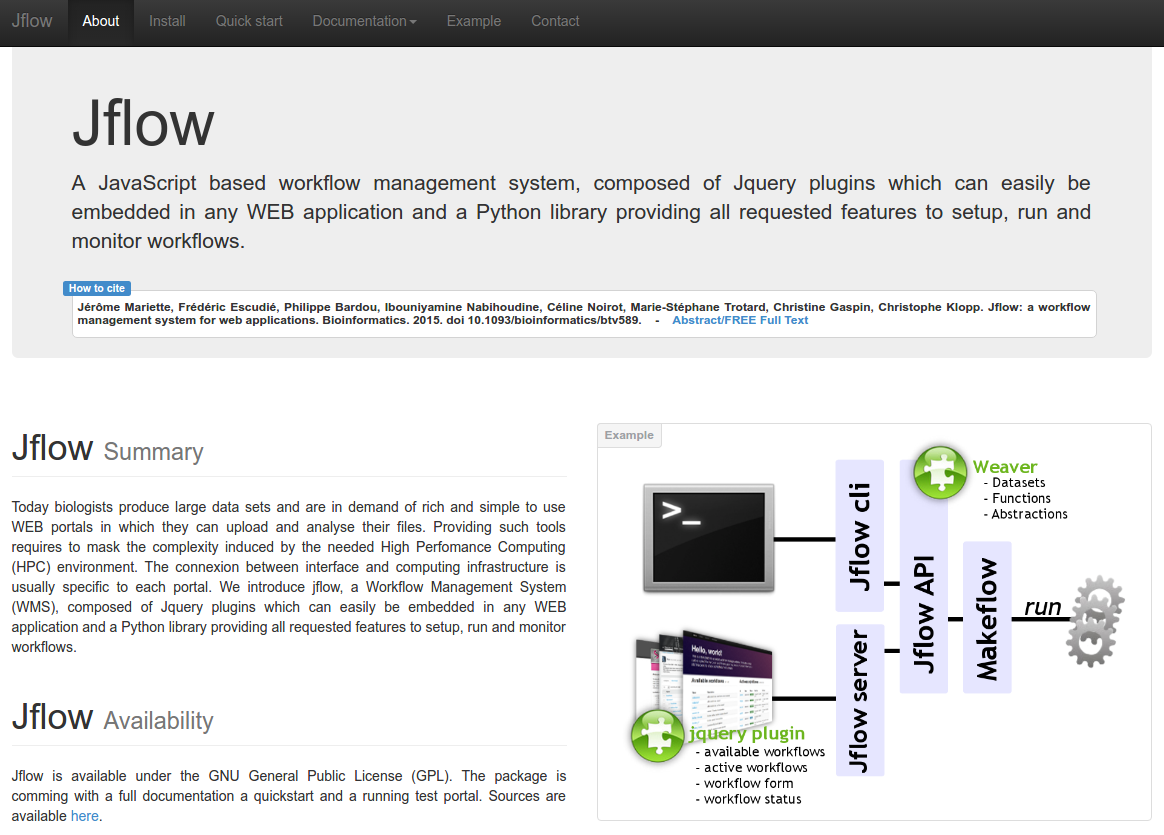 Description: Building rich WEB environments aimed at helping scientists analyse their data is a common trend in bioinformatics. These applications are often specialized WEB portals or generic Workflow management systems (WMS). The first class provides multiple services and analysis tools in an integrated interface for a specific experiment or data type. Quite often these systems hide the processing steps in the back-office. The second class is mainly focused on workflow creation and provides a rather poor end-user interface but enable to combine tools and data sources as desired. Jflow is a fully scalable WMS that can easily be embedded in any WEB site, providing all WMS features and benefits to your project.
Description: Building rich WEB environments aimed at helping scientists analyse their data is a common trend in bioinformatics. These applications are often specialized WEB portals or generic Workflow management systems (WMS). The first class provides multiple services and analysis tools in an integrated interface for a specific experiment or data type. Quite often these systems hide the processing steps in the back-office. The second class is mainly focused on workflow creation and provides a rather poor end-user interface but enable to combine tools and data sources as desired. Jflow is a fully scalable WMS that can easily be embedded in any WEB site, providing all WMS features and benefits to your project.
Availability: Jflow code is freely available on INRAE Forge.
Publication: Jérôme Mariette, Frédéric Escudié, Philippe Bardou, Ibouniyamine Nabihoudine, Céline Noirot, Marie-Stéphane Trotard, Christine Gaspin, Christophe Klopp. Jflow: a workflow management system for web applications. Bioinformatics. 2015. doi 10.1093/bioinformatics/btv589. see
jvenn
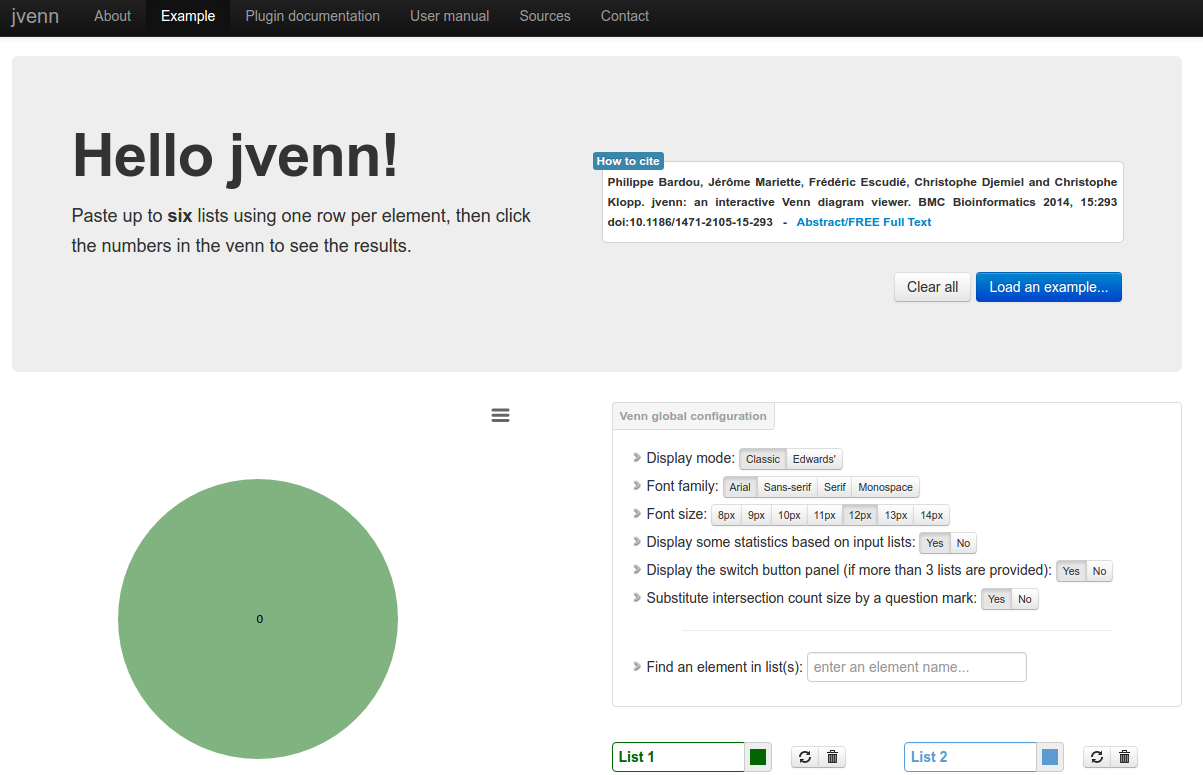 Description: jvenn is a plug-in for the jQuery Javascript library. It is a flexible tool, based upon the venny tool first developed by J.C Oliveros (Oliveros, J.C (2007) VENNY, An integrative tool for comparing lists with Venn Diagrams). Key features: hanldes up to 6 classes venn diagram, allows to display Edwards-Venn diagram, is easily integrable within your own web site, allows to provide the data from 3 different ways (lists/intersection counts/count lists), control the click callback function, provides statistic charts based on input data, search for elements, exports the venn diagram to PNG and SVG, exports lists to CSV.
Description: jvenn is a plug-in for the jQuery Javascript library. It is a flexible tool, based upon the venny tool first developed by J.C Oliveros (Oliveros, J.C (2007) VENNY, An integrative tool for comparing lists with Venn Diagrams). Key features: hanldes up to 6 classes venn diagram, allows to display Edwards-Venn diagram, is easily integrable within your own web site, allows to provide the data from 3 different ways (lists/intersection counts/count lists), control the click callback function, provides statistic charts based on input data, search for elements, exports the venn diagram to PNG and SVG, exports lists to CSV.
Availability: The plugin code and its documentation is freely available on INRAE Forge. A running version of the plugin is also available from our WEB site.
Warning: Please note that we are not adding any new features to this software.
Publication: Philippe Bardou, Jérôme Mariette, Frédéric Escudié, Christophe Djemiel and Christophe Klopp. jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 2014, 15:293 doi:10.1186/1471-2105-15-293. see
RNAbrowse
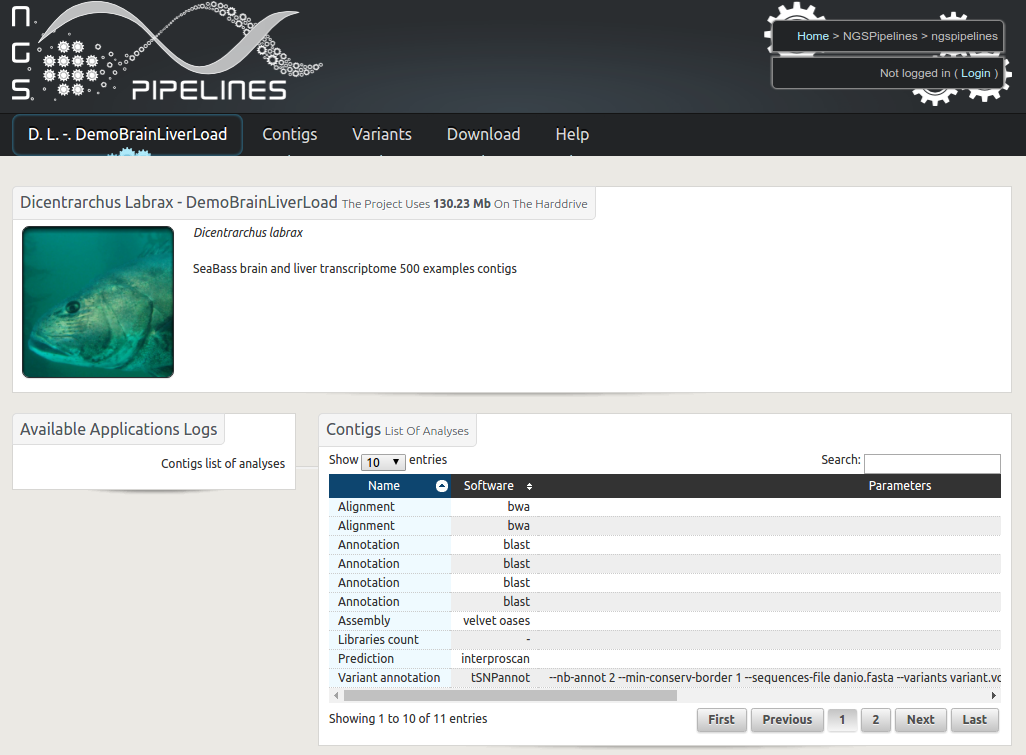 Description : Transcriptome analysis based on a de novo assembly of next generation RNA sequences is now performed routinely in many laboratories. The generated results, including contig sequences, quantification figures, functional annotations and variation discovery outputs are usually bulky and quiet diverse. RNAbrowse is an user oriented storage and visualisation environment permitting to explore the data in a top-down manner, going from general graphical views to all possible details. The software package is based on biomart, easy to install and populate with local data.
Description : Transcriptome analysis based on a de novo assembly of next generation RNA sequences is now performed routinely in many laboratories. The generated results, including contig sequences, quantification figures, functional annotations and variation discovery outputs are usually bulky and quiet diverse. RNAbrowse is an user oriented storage and visualisation environment permitting to explore the data in a top-down manner, going from general graphical views to all possible details. The software package is based on biomart, easy to install and populate with local data.
Availability: The software package is available under the GNU General Public License (GPL) and can be downloaded from INRAE Forge (workflow rnaseqdenovo).
Publication: Mariette J, Noirot C, Nabihoudine I, Bardou P, Hoede C, Djari A, Cabau C, Klopp C. (2014) RNAbrowse: RNA-Seq De Novo Assembly Results Browser. PLoS ONE 9(5): e96821. doi: 10.1371/journal.pone.0096821. see
Published instance: Magnanou et al, 2014; Hinaux et al, 2013; Riviere et al, 2015
NG6
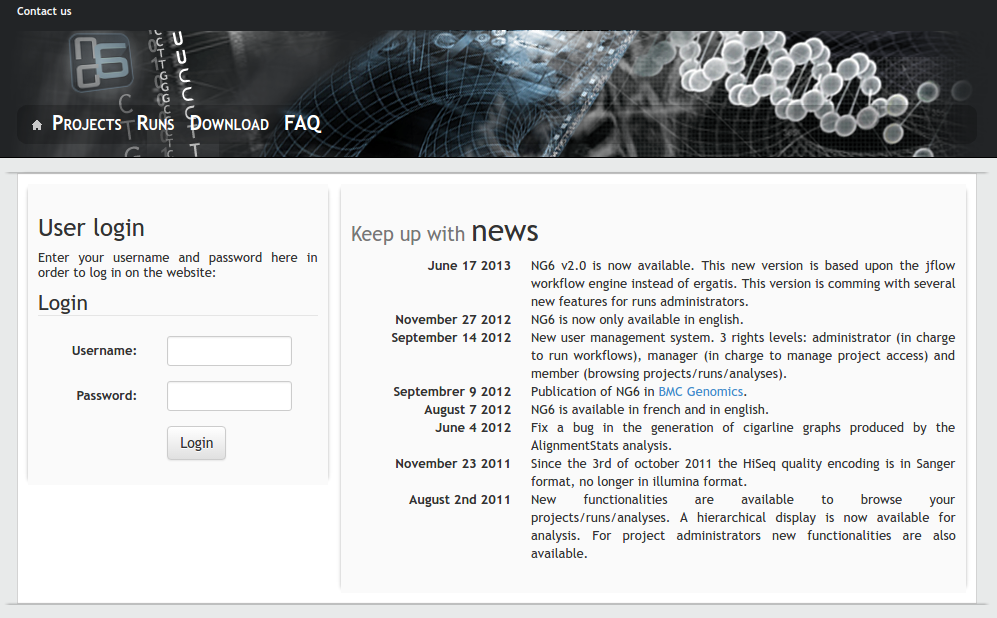 Description: The platform works in tight collaboration with the GeT sequencing platform for the management and the analysis of data produced by their Roche 454 and Illumina HiSeq sequencers. NG6 is an extensible sequencing provider oriented LIMS. It includes read quality control and first level analysis processes which ease the data validation made jointly by the sequencing facility staff ant the end-users. It provides a secured user-friendly interface to visualize and download the raw sequences files and the analysis results.
Description: The platform works in tight collaboration with the GeT sequencing platform for the management and the analysis of data produced by their Roche 454 and Illumina HiSeq sequencers. NG6 is an extensible sequencing provider oriented LIMS. It includes read quality control and first level analysis processes which ease the data validation made jointly by the sequencing facility staff ant the end-users. It provides a secured user-friendly interface to visualize and download the raw sequences files and the analysis results.
Availability: The NG6 code is freely available on INRAE Forge. To ease the installation, the package and all its dependencies are also available as a virtual machine. Installing and maintaining the system would require expertise in Linux system administration.
Publication: Mariette J, Escudie F, Allias N, Salin G, Noirot C, Thomas S, Klopp C. NG6: Integrated next generation sequencing storage and processing environment. BMC Genomics, 2012 13:462. see
RNAspace
 Description: RNAspace is a platform which aims at providing an integrated environment for non-coding RNA annotation. The increasing number of ncRNA discovered since 2000 and the lack of user friendly tools for finding and annotating them, have made necessary to propose to biologists an in silico environment allowing structural and functional annotations of these molecules with regard to available protein genes annotation environments. RNAspace makes available a variety of ncRNA gene finders and ncRNA databases as well as user-friendly tools to explore computed results including comparison, visualization and edition of putative RNAs. RNAspace also allows to export putative RNAs in various formats.
Description: RNAspace is a platform which aims at providing an integrated environment for non-coding RNA annotation. The increasing number of ncRNA discovered since 2000 and the lack of user friendly tools for finding and annotating them, have made necessary to propose to biologists an in silico environment allowing structural and functional annotations of these molecules with regard to available protein genes annotation environments. RNAspace makes available a variety of ncRNA gene finders and ncRNA databases as well as user-friendly tools to explore computed results including comparison, visualization and edition of putative RNAs. RNAspace also allows to export putative RNAs in various formats.
Availability: RNAspace is an open source project. It is developed in Python. It is copyrighted with the GNU General Public License, and is free (in the GNU sense) for all to use, and is in constant development. RNAspace is hosted at Sourceforge. It is also available as a web server at rnaspace.org.
Publication: Marie-Josée Cros, Antoine de Monte, Jérôme Mariette, Philippe Bardou, Benjamin Grenier-Boley, Daniel Gautheret, Hélène Touzet, and Christine Gaspin. RNAspace.org: an integrated environment for the prediction, annotation and analysis of ncRNA. RNA 2011 17 (11) see