DRAP
 Description: Short read RNASeq de novo assembly is a well established method to study transcription of organisms lacking a reference genome sequence. Available software packages such as Trinity and Oases have proven to be able to build high quality contigs from short reads. But there is still room for improvement on different points such as i) compactness: they often produce different contigs which are included in one another or overlapping one another, ii) chimerism: the contigs contain different kinds on chimera such as duplicated open reading frames, iii) substitution, insertion, deletion errors: the consensus sequences build by the assembler contain errors which can be partly corrected using the read alignments.
Description: Short read RNASeq de novo assembly is a well established method to study transcription of organisms lacking a reference genome sequence. Available software packages such as Trinity and Oases have proven to be able to build high quality contigs from short reads. But there is still room for improvement on different points such as i) compactness: they often produce different contigs which are included in one another or overlapping one another, ii) chimerism: the contigs contain different kinds on chimera such as duplicated open reading frames, iii) substitution, insertion, deletion errors: the consensus sequences build by the assembler contain errors which can be partly corrected using the read alignments.
Availability: DRAP code is freely available on the forgemia.
Partners: team SIGENAE.
FROGS
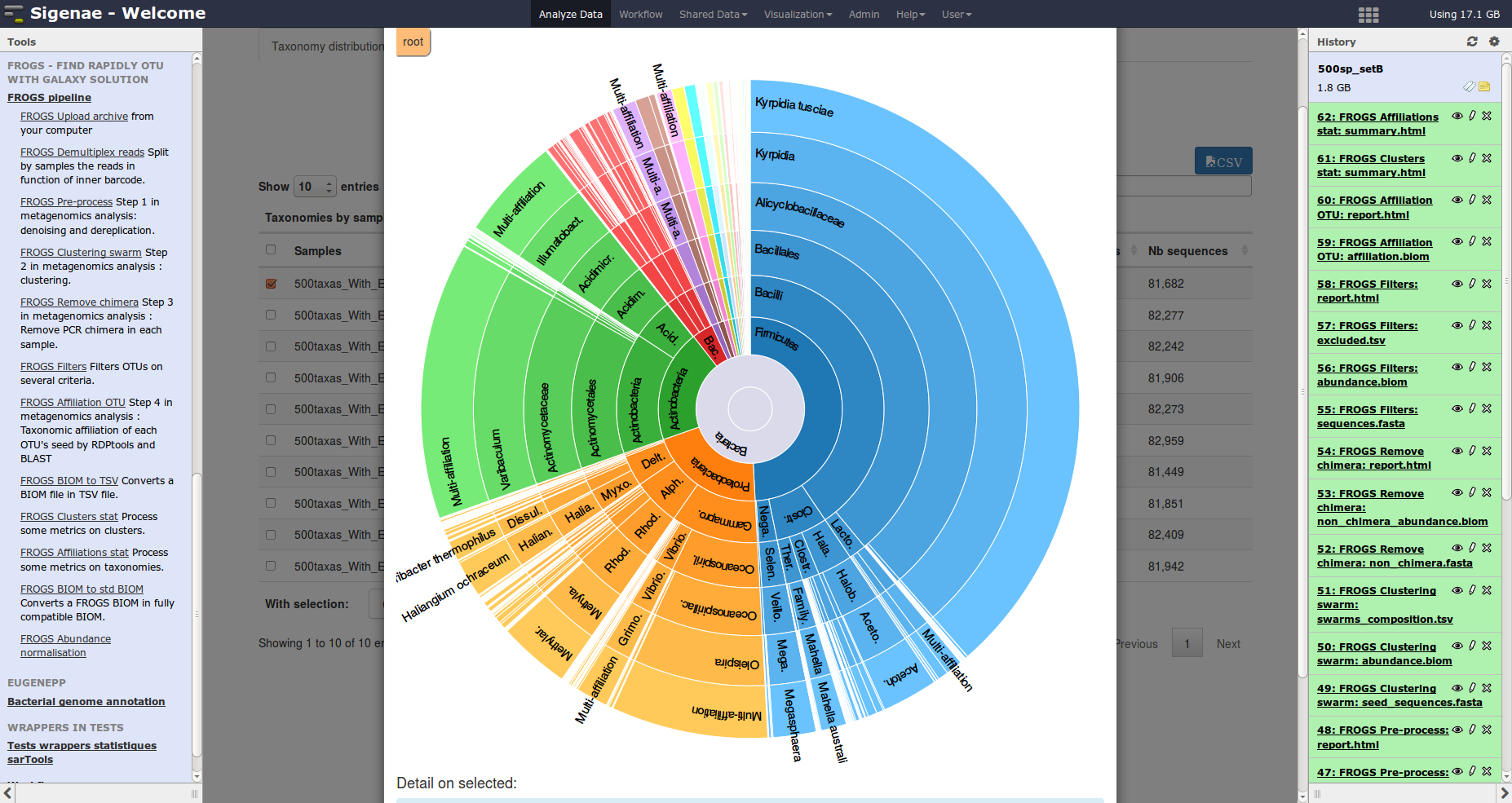 Description: FROGS is a galaxy/CLI workflow designed to produce an OTU count matrix from high depth sequencing amplicon data. This workflow is focused on: i) user-friendliness with the integration in galaxy and lots of rich graphic outputs, ii) accuracy with a clustering without global similarity threshold, the management of multi-affiliations and management of separated PCRs in the chimera removal step, iii) speed with fast algorithms and an easy to use parallelisation, iv) scalability with algorithms designed to support the data growth.
Description: FROGS is a galaxy/CLI workflow designed to produce an OTU count matrix from high depth sequencing amplicon data. This workflow is focused on: i) user-friendliness with the integration in galaxy and lots of rich graphic outputs, ii) accuracy with a clustering without global similarity threshold, the management of multi-affiliations and management of separated PCRs in the chimera removal step, iii) speed with fast algorithms and an easy to use parallelisation, iv) scalability with algorithms designed to support the data growth.Availability: FROGS code is freely available on GitHub.
Partners: team NED (GenPhySE), SIGENAE and Migale.